|
|
|
|
05/26/25 |
|
|
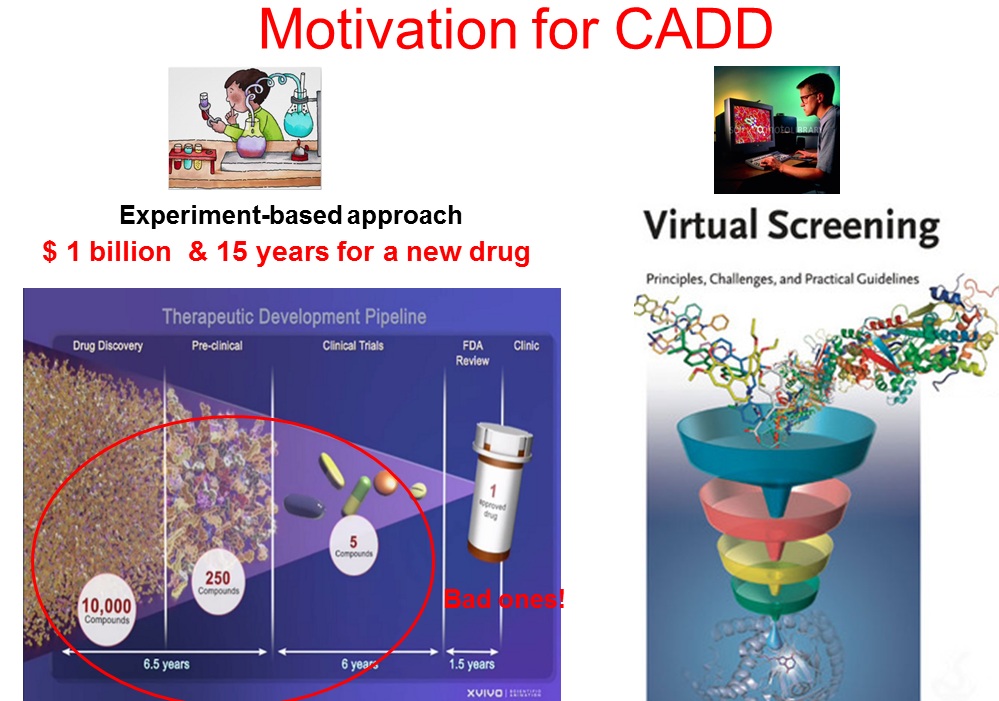
My research interests are in the area of computer-aided drug designs (CADD). CADD has a great potential to hasten the process of drug development and reduce the developing cost, just like its counter-part in developing mechanic and electronic devices (e.g. Boeing latest dreamliner and iPad are designed and optimized in computers before actual manufacturing.). CADD is a technique that is revolutionizing the pharmaceutical industry.
Bioinformatics: Wu’s lab is pioneering a bioinformatics tool—the substitution-to-mutation rate ratio (C ⁄ μ) test—to quantify the fitness effects of mutations directly from genomic sequence data. Applied to viral genomes, the method has given rise to the Near-Neutral Selectionist Theory (NNST), a novel framework that clarifies the molecular evolution of SARS-CoV-2 and other pathogens while pinpointing genomic hotspots vital for designing both current and next-generation vaccines and therapeutics to combat infectious diseases. Molecular modeling, docking and molecular dynamics simulation are the key tools in CADD. In particular, molecular dynamics (MD) is a powerful tool to probe the structure and dynamics of biomolecules with high temporal and spatial resolution. Because the structure and dynamics of biomolecules (DNA, proteins etc.) determine their biological functions, fast and accurate structure predictions using MD simulations will not only better our understanding of biology, but also lead to powerful application in CADD. The predicting power of MD simulations is determined by two factors: the accuracy of force field (a parameter set for describing physical interactions among atoms) and the completeness of conformation space sampling. I have been active in improving these two factors by making significant contributions in developing a popular AMBER protein force field (ff03, over 3000 citations) and in implementing high-performance sampling algorithms. My research focuses on two directions: improve predictive power of MD simulations and use MD simulations for various biomedical applications. Computing resources: Hardware 1). 15 workstations 2). 5 GPU workstations (GTX 980) 3). Local 16-node cluster with 288 cores (Intel Xeon E5-2650v2, 2.6 GHz) 4). 42-node HPC cluster with 1200 cores ( Intel Xeon E5-2650v2, 2.3 GHz), shared with Professors Bouaynaya, Polikar, Sukumaran etc. 5). ~1000,000 SUs per year on national super computers from XSEDE. 6). Allocation from Beijing Computational Science Research Center.
Software 1). Schroedinger 2). AMBER 3). GAUSSIAN 09
Research projects: My lab has been carrying out a large number of CADD projects in collaborating over thirteen research groups including nine groups at Rowan University and four groups from the other institutes. These CADD projects aim to 1) Develop novel cancer drugs that target DNA-quadruples, transporter and kinase. 2) Develop novel analgesic and anti-addition agents that target GPCR membrane receptors. 3). Develop novel anti-virus drugs against herpes virus entry and HIV integration. 4). Develop diabetes drug that simultaneously targets inflammation. The collaborations has identified the lead compounds, and produced multiple posters publications and grant proposals. The selected collaborations are discussed below:
Professor Claude Krummenacher, Biological Sciences and Molecular & Cellular Sciences Discovery of novel agent to prevent herpes infection
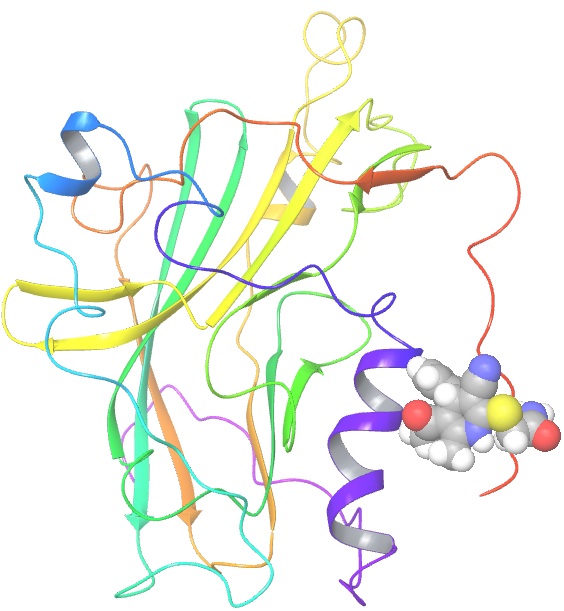
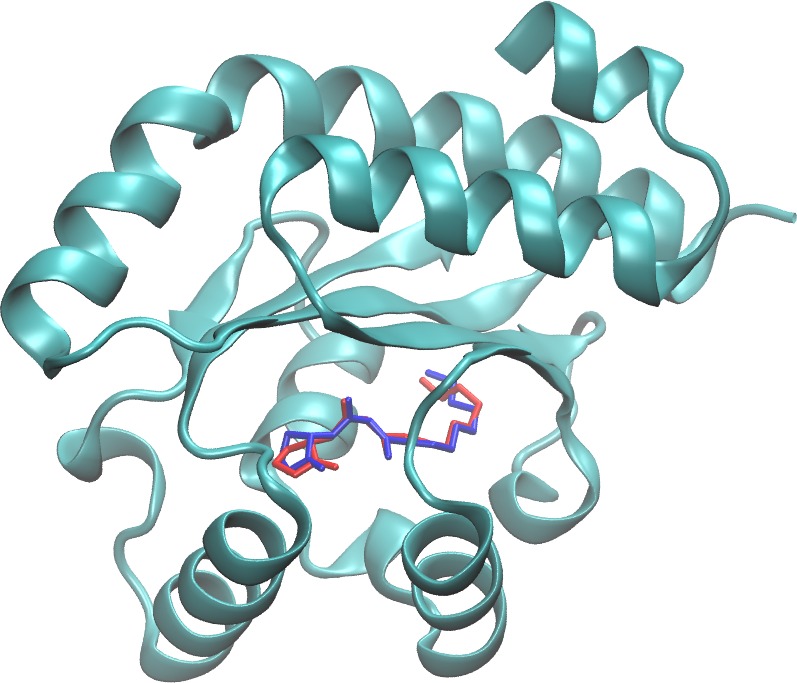
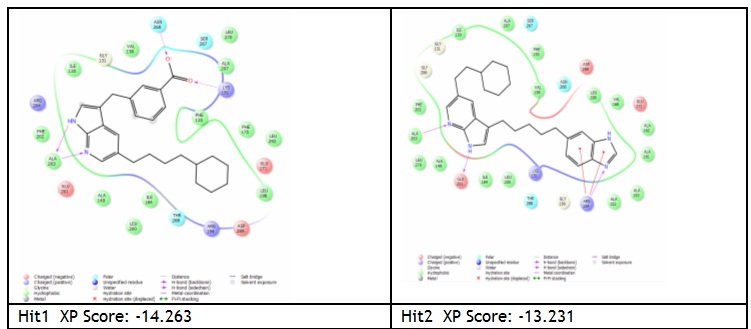
Herpes simplex virus type 1 (HSV-1) is the cause of cold sores and affects 57% of people in the United States. It is a lifelong infection, for which there is no cure and no vaccine. After a primary infection, HSV-1 remains latent in sensory neurons until reactivated by a stressor. Currently, prescription medications such as Zovirax and Valtrex are used to prevent/manage reactivation symptoms. However, there are a growing number of clinical isolates that are resistant to these drugs which creates the need for novel treatments/preventive options for HSV-1. We aim at designing drugs that would prevent primary infection and limit reactivation, by blocking virus entry into cells. The drugs in this study were identified in a small compound library using computer modeling of their binding to a key location of the viral entry glycoprotein gD. We anticipate that binding of these drugs to gD will block interaction with the cell receptor and prevent virus infection ten compounds were obtained and are being tested for their ability to inhibit infection of cells and to prevent gD-receptor interaction in vitro, as tested by ELISA. The Wu group screened the Zinc database (~2.3 millions compounds) using a herpes glycoprotein structure to isolate small molecules that could block HSV infection. From the top 20 hits, the Krummenacher purchased about 10 of them (not all were commercialized). They tested them for toxicity on cells, the ability to limit infections, and to interfere with the viral glycoprotein binding to its receptors. Two compounds showed promising inhibitory activity. We are filing a disclosure for the activity of these drugs.
gD protein with a docked ligand
Professor Subash Jonnalagadda Chem &Biochem and Molecular & Cellular Sciences Professor Manoj Pandey Dept. of Biomedical Sciences of Cooper Medical School
Several salinomycin analogs have been synthesized in Dr. Jonnalagadda lab. The preliminary biological data shows promising activity against several cancer cell lines. To illuminate the mechanism of action, we conducted molecular docking and simulations to identify the drug targets. Dr. Pandey lab will carry out biochemical assay and cancer cell line experiments to verify the prediction from our calculations. With solid preliminary data, we will apply for NIH/industry grants to optimize/develop this class of novel anti-cancer drugs.
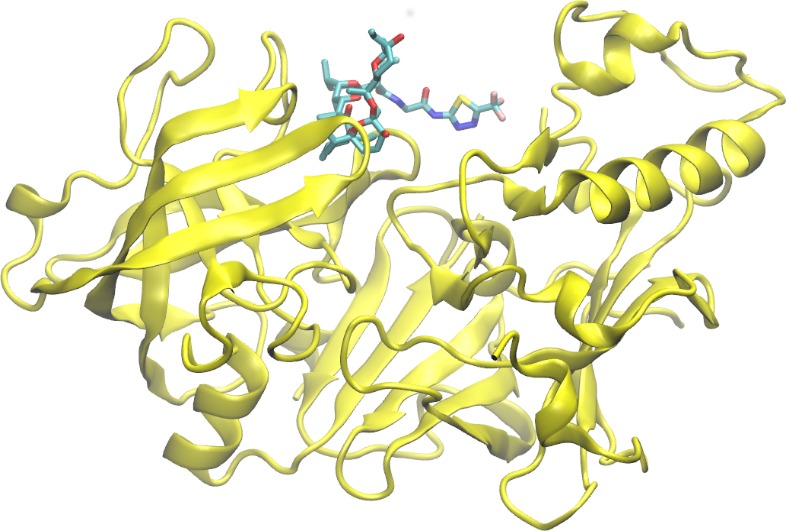
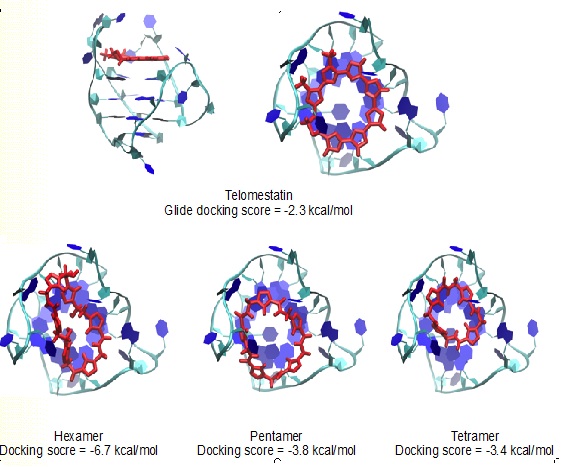
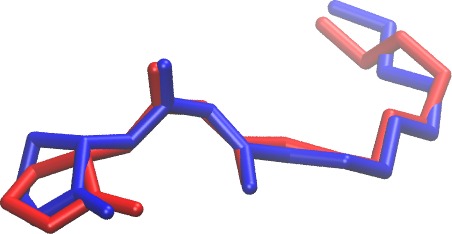
Professor Gustavo Moura-Letts Chem &Biochem and Molecular & Cellular Sciences DEVELOPMENT OF NOVEL TELOMERASE INHIBITORS FOR THE TREATMENT OF OVARIAN CANCER Ovarian cancer is the deadliest cancer that targets the female reproductive system and is ranked the fifth deadliest cancers affecting women in the United States. The unlimited proliferative potential of cancer cells primarily depends on telomere maintenance. In ovarian tissues, telomerase is commonly absent in normal ovarian surface epithelium and premalignant lesions but up-regulated in over 95% of ovarian carcinomas. Recent efforts have shown that stabilizing the telomeric G-quadruplexes can inhibit telomerase activity, thus the development of G-quadruplex ligands small-molecule inhibitors as anticancer drug candidates is the current focus of the cancer therapeutic field. The Moura-Letts group have developed an efficient synthetic method for the production of polyisoxazoles (tetramers, pentamers and hexamers) from easily accessible substituted isoxazole and isoxazoline building blocks. These molecules are poised to mimic Telomestatin properties as telomerase inhibitors. Furthermore, we believe that polyisoxazoles have better drug-like properties and have the high potential to become actual ovarian cancer therapeutics. The Wu group will validate the hypothesis by assessing their binding mechanism to key intramolecular G-quadruplexes using molecular docking and all-atom molecular dynamics simulations. The simulations will obtain the high-resolution structure of G-quadruplex-ligand complexes and uncover their recognition mechanisms. These efforts will provide a virtual library of polyisoxazole scaffolds with high binding affinities. This second-generation library will be synthesized (12 to 16 analogues) using our proposed synthetic method. The resulting targets will then be screened using in vitro cell-based assays to identify the best candidate telomerase inhibitor.
Docking of Telomestatin and polysixazole to an intramolecular (3+1) human telomeric G-quadruplex.
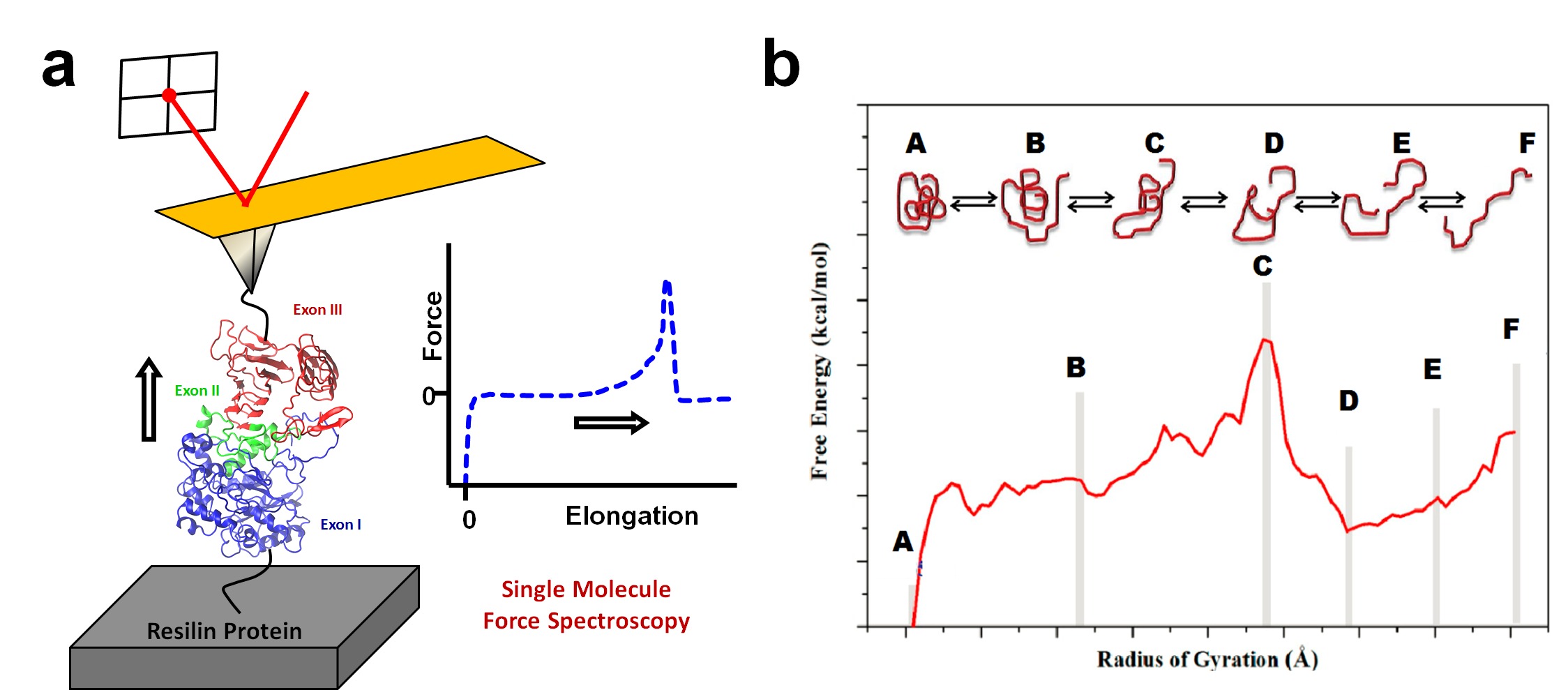
Professor Xiao Hu, Department of Physics and Astronomy, Rowan University RUI: Unraveling the Structure-Activity Relationships of Resilin Biomaterials by Joint Experimental-Computational Approaches Resilin, known as a “super elastic rubber”, has drawn intensive research interests for its striking energy transduction and storage properties. However, due to lack of atomic structures of this protein and its constituent domains, no models with atomic details are available up to date to enable an in-depth understanding of the energy transductions and storage mechanism in resilin. In our current study, with model peptides, we explore the structures of the high-hydrophobicity sequence and the repeats of the Exon III domain of Drosophila melanogaster resilin at the atomic level with combinations of computational and experimental techniques. Bioinformatics method predicts dominant β-sheet and β-turn structures for the above respective sequences, which is consistent with earlier and present experimental acquisitions. Molecular dynamics (MD) simulations with the explicit solvent model further confirm these structural features. The peptide secondary and tertiary structures, and peptide-water interactions are investigated in details with simulations, and the results are compared to experimental measurements from Fourier Transform Infrared Spectroscopy (FTIR) and Circular Dichroism (CD). The present study offers an atomistic view of resilin and elastomeric proteins in general where β-turn-related structures serve as fundamental units of the structure and elasticity. Meanwhile, the high-hydrophobicity sequence of the Exon III domain is proposed to play a critical role in resilin energy storage and transduction.
a) Experimental approach to measure expansion-contraction transition of resilin molecules using the Single Molecule Force Spectroscopy method through AFM. (b) Correlated computational free energy profile for the expansion-contraction transition of resilin. PMF profiles along the reaction coordinate of the radius of gyration will be obtained from umbrella sampling with weighted histogram analysis method (WHAM).
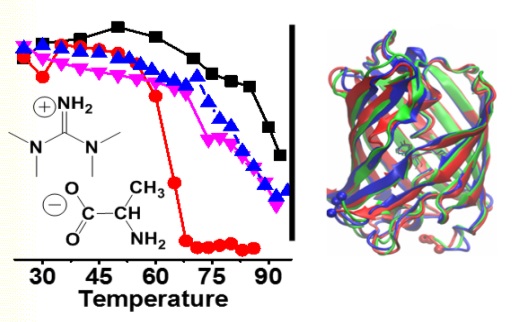
Professors Timothy Vaden and Gregory Caputo, Dept. of Chemistry and Biochemistry, Rowan University Project Background: Ionic liquids (ILs) have recently found a variety of novel biomedical and biochemical applications. ILs have been used to solubilize biomaterials, inhibit or enhance enzyme activities, enhance antibiotic effectiveness or act as novel antimicrobial compounds, act as drug delivery systems, and even function as cosmetics formulations ingredients. A natural question arises when studying ILs and biological systems regarding the nature of interactions between ILs and biomolecules. This question is central to the development of safe, useful, and effective ILs for biomedical applications. Hence, numerous recent investigations have reported protein – IL interactions and protein structures and stabilities in the presence of ILs in aqueous solution, and have provided models of protein interactions with ILs from molecular dynamics simulations and spectroscopic characterization. Most studies have focused on simple helical proteins such as BSA, lysozyme, myoglobin, etc. in isolated aqueous solutions, and have focused on equilibrium experimental conditions. We are interested in understanding how ILs interact with proteins different of secondary structure motifs (in β-sheet, β-barrel, and other complex structures), in membranes or other volume-restricted physiological environments, and how the ILs affect protein dynamics. Project Description: The project will combine the biophysical chemistry experience of three groups in the Dept of Chemistry and Biochemistry at Rowan University in order to understand the molecular interactions between ILs and -sheet, -barrel, and membrane proteins, how these interactions affect the protein conformational dynamics, and how these effects differ between proteins in dilute buffer solutions and in membranes or confined environments. Dr. Gregory Caputo (biochemist) will synthesize and purify proteins from genetically coded E. Coli or other bacterial sources. Dr. Timothy Vaden (biophysical chemist) will prepare ILs, prepare proteins in various samples and environments, and characterize proteins using IR spectroscopy, CD spectroscopy, and mass spectrometry. Dr. Vaden will also perform thermodynamics (protein unfolding) and kinetics/dynamics measurements using fluorescence correlation spectroscopy (equipment at a different university). Dr. Chun Wu (computational chemist) will run molecular dynamics simulations to probe the specific IL-protein interactions, and quantify the atomic and molecular-level pictures in terms of protein structures, stabilities, and dynamics. Most equipment is in place and the budget will provide for faculty summer salaries, student salaries, consumable materials and equipment (test tubes, etc), and some minor spectroscopy equipment (eg, a time-resolved fluorimeter). Project Aims and Goals: The project aims to use experiments and simulations to understand the effects of ILs in solution on various proteins of “real-world” applicability in different environments. The goals are (1) to develop an understanding of how ILs affect protein structures and conformational stabilities; (2) to develop novel ILs that would be biocompatible and have “designed” effects of proteins in physiologically-relevant environments; and (3) to quantify the differences between protein-IL interactions in solution and protein-IL interactions in cell membranes.
Broader Impacts:
Rowan University (especially the Dept. of Chemistry
and Biochemistry) is a primarily undergraduate research institution and the
project will train undergraduate students. We also hope to develop the
investigations into laboratory experiences for our upper-level physical
chemistry, biophysical chemistry, and molecular modeling courses. These
experiences would demonstrate the power of combining molecular simulations
with biochemistry and spectroscopy experiments to understand the
interactions involving a protein at the molecular level for “novel” systems.
Publication: Borrell, K. L.; Cancglin, C.; Stinger, B. L.; DeFrates, K. G.; Caputo, G. A.; Wu, C.*; Vaden, T. D., An Experimental and Molecular Dynamics Study of Red Fluorescent Protein mCherry in Novel Aqueous Amino Acid Ionic Liquids. The Journal of Physical Chemistry B 2017, 121 (18), 4823-4832.
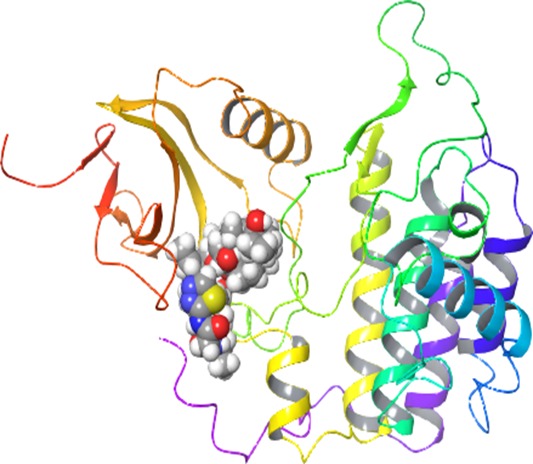
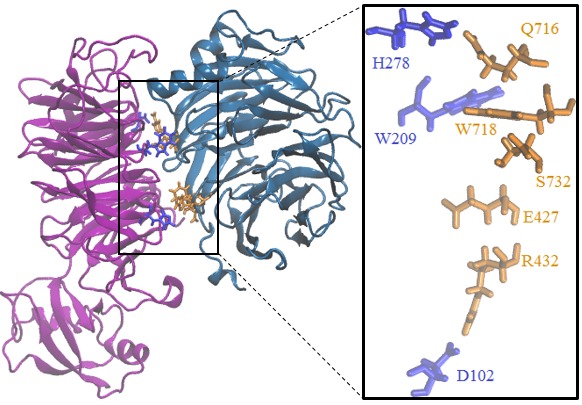
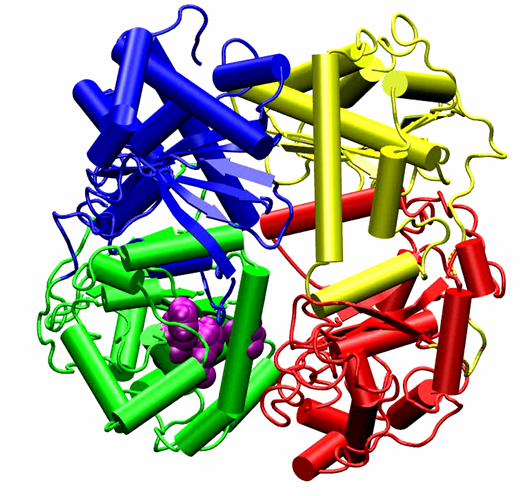
Professor Dimitri Pestov, Department of Cell Biology, Rowan University-School of Osteopathic Medicine Targeting ribosome biogenesis as a new strategy to enhance cancer chemotherapy. Chemotherapy remains the mainstay of cancer treatment, but it is invariably associated with serious side effects. In addition, cancer cells often develop resistance to chemotherapeutic drugs, leading to treatment failure. Thus, new approaches to chemotherapy are needed for improving clinical outlook and quality of life in cancer patients. The goal of the proposed study is to develop a new class of pharmacological agents that will make chemotherapy safer and more effective. Our recent studies identified cellular proteins involved in the assembly of ribosomes as a promising molecular target for making normal cells in the body more resistant to anticancer drugs. We found that by temporarily disabling activities of one of these proteins called Bop1, we could protect cultured cells from treatments with high doses of chemotherapeutic drugs camptothecin and metothrexate that would normally be lethal for these cells. Furthermore, this protection requires the controls genome stability in normal cells but is often lost in human cancers. Because cytoprotection through Bop1 only works in noncancerous, p53-positive cells, resistance of normal cells can be selectively increased while targeting p53-negative cancer cells with chemotherapeutic drugs. To start the development of novel pharmacological agents based on these findings, we will first use advanced computational tools to pinpoint parts of the Bop1 molecule that could be targeted via small-molecule chemical compounds. The predicted druggable surfaces in Bop1 will be validated by introducing point mutations into Bop1 and biochemical assays. Next, we will virtually dock millions tens of thousands of small molecules to the selected parts of the Bop1 structure to identify candidate inhibitors. By using in silico drug discovery at the initial stages, we expect to dramatically reduce the time and costs necessary to identify chemical compounds that will inhibit Bop1 function. In the future research, these pharmacological candidates will be used to test cytoprotection-enhanced anticancer drug treatments in animal models, moving us a step closer to improved chemotherapeutic interventions in human cancers.
Homomolgy model of mouse Wdr12-Bop1 (purple-blue) and some key Bop1 residues for mutations.
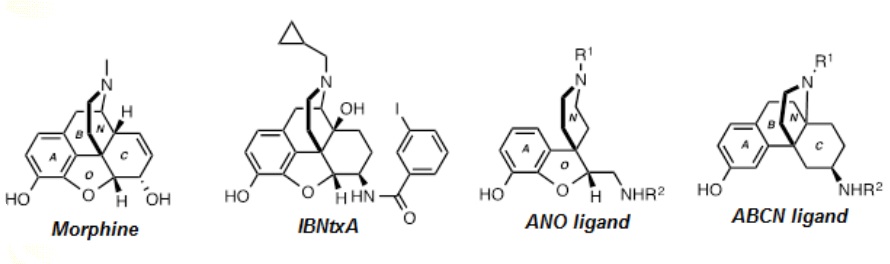
Professors Moura-Letts Gustavo and Thomas M. Keck, Chemistry & Biochemistry, Rowan Univ. Understanding the Consequences of 6TM Splice Variants in MOR-1 Ligand Binding: Towards Novel Analgesics Opioid drugs are a critical class of medications to treat acute and chronic pain, a serious and costly public health issue. The prototypical opioid analgesic morphine (Figure 1) is on the WHO Model List of Essential Medicines, the most important medications needed in a basic health system. However, medical use of opioid analgesics is limited by critical side effects including sedation, constipation, abuse liability and respiratory depression. There is pressing need for the development of novel opioid analgesics with more precisely targeted mechanisms of action. Opioid analgesics induce their therapeutic effects primarily through the activation of the μ opioid receptor (MOR-1), found on a wide variety of central and peripheral neurons. MOR-1 is a seven transmembrane (7TM) G protein-coupled receptor (GPCR). Many splice variants of the MOR-1 gene, OPMR1, in rodents and humans are actively expressed and appear to physiologically mediate analgesic responses. Notably, a subset of truncated variants of OPMR1 have altered sensitivity to morphine and other classical opioid analgesics in vitro and are predicted to contain only six TM domains (6TM). Recent studies have identified the novel analgesic IBNtxA (Figure 1) as a preferential agonist of 6TM variants, providing potent analgesia with reduced side effects compared to morphine. These results raise the hope that it might be possible to dissociate opioid analgesia from side effects and abuse liability. Thus, targeting 6TM variants may yield important new analgesics in the future. However, there is no high-resolution receptor/complex structure of the 6TM MOR variants, and it is not clear how the receptor truncation alters the ligand binding properties of these receptors. Although IBNtxA is a promising drug candidate, its high molecular complexity derived from its substituted oxymorphone alkaloid core may prevent it from further development as a clinical drug. Our overall goal is to develop novel ligands selective toward the human MOR-1 6TM splice variants (hMOR-1G1 and hMOR-1G2) using combined computational and experimental approaches. Based on preliminary docking data, two simplified scaffolds of IBNtxA have been identified as initial lead structures. However, the number of possible ligands is prohibitively large for synthesis and in vitro screening. We will apply our computational screening to reduce the number to ~50, which will then synthesized and tested in vitro.
Figure 1 Chemical structures of morphine, IBNtxA and proposed novel scaffolds.
Professor Lark J. Perez, Chemistry and Biochemistry, Rowan University Development of a novel series of non-natural triaryl agonists and antagoinsts of the pseudomonas aeruginosa LasR quorum sensing receptor Bacterial chemical communication, through a process called quorum sensing (QS), plays a central role in infection in numerous bacterial pathogens. Quorum sensing in Pseudomonas aeruginosa employs a series of small molecule r eceptors including the master QS regulator, LasR. In this study we investigate a non-natural triaryl series of LasR ligands using a combination of structure activity relationship studies and computational modeling. These studies have enabled the identification of key structural requirements for ligand binding and have revealed a new strategy for inducing the therapeutically relevant antagonism of LasR. Publication: Capilato JN, Philippi SV, Reardon T, McConnell A, Oliver DC, Warren A, Adams JS, Wu C, Perez LJ*(2017) “Development of a novel series of non-natural triaryl agonists and antagoinsts of the Pseudomonas aeruginosa LasR quorum sensing receptor”, Bioorg Med Chem. 25(1):153-165
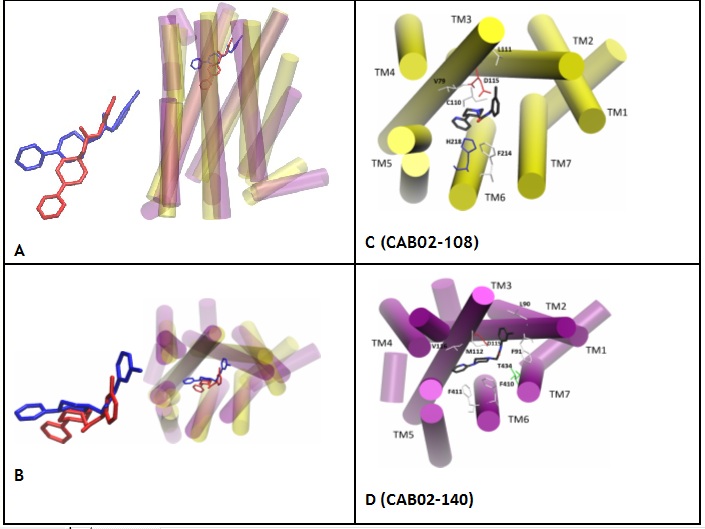
Professor Thomas M. Keck, Chemistry and Biochemistry, Rowan University Development and binding of novel D4 receptor-selective compounds as agonists used to study neuropsychiatricdisorders The dopamine D4 receptor (D4R) is a synaptic GPCR, coupled to Gai/o, primarily localized in the prefrontal cortex and hippocampus wherein D4R signaling can modulate memory, cognition, attention, and executive function. Thus, pharmacological modulation of D4R signaling may be useful to treat conditions such as attention-deficit/hyperactivity disorder, Alzheimer’s disease, and drug addiction: D4R agonism may improve cognition while antagonism may reduce drug-taking behavior. This project seeks to design novel D4R-targeted therapeutics for preclinical evaluation. Using the classical D4R partial agonist A-412997 as a structural template, analogues were synthesized and evaluated for in vitro receptor affinity and activity in transfected D4R-expressing HEK293 cells. Some compounds were found to bind with higher affinity to D4R than A-412997, with greater selectivity over related D2R and D3R. The highest affinity compounds were potent partial agonists in cAMP and beta-arrestin functional assays. Using in silico modeling techniques, a D4R model was created from the X-ray crystal structure of D3R allowing for the analysis of likely binding poses and predictions of the binding affinities of a second generation novel library. Refinement of this in silico method could lead to the capability to perform high-throughput screening of thousands of compounds for affinity at D4R, as well as the development of new pharmacotherapies for the treatment of neuropsychiatric disorders.
Binding pose comparison between CAB02-108/A412997 and CAB02-140. A-B: CAB02-108(red)/D4(yellow) CAB02-140(blue)/D4(purple) C-D: ligand (C: black, N: blue, O:red), residue (hydrophobic: white, basic: blue, acidic: red, hydrophilic, green).
Professor Pui-Kai (Tom) Li, Division of Medicinal Chemistry and Pharmacognosy, The Ohio State Univ. Development MLK(Mixed Lineage Kinase) inhibitor as anti-cancer agents Mixed lineage kinases (MLKs) are members of the mitogen-activated protein kinase kinase kinase (MAP3K) family and are reported to activate MAP kinase pathways. There have been at least 9 members of the MLK family identified to date, although the physiological functions of all the family members are yet unknown. However, MLKs in general have been implicated in neurodegenerative diseases, including Parkinson and Alzheimer diseases. Recent reports suggest that some of the MLK members could play a role in cancer via modulating cell migration, invasion, cell cycle, and apoptosis. This review article will first describe the biology of MLK members and then discuss the current progress that relates to their functions in cancer.
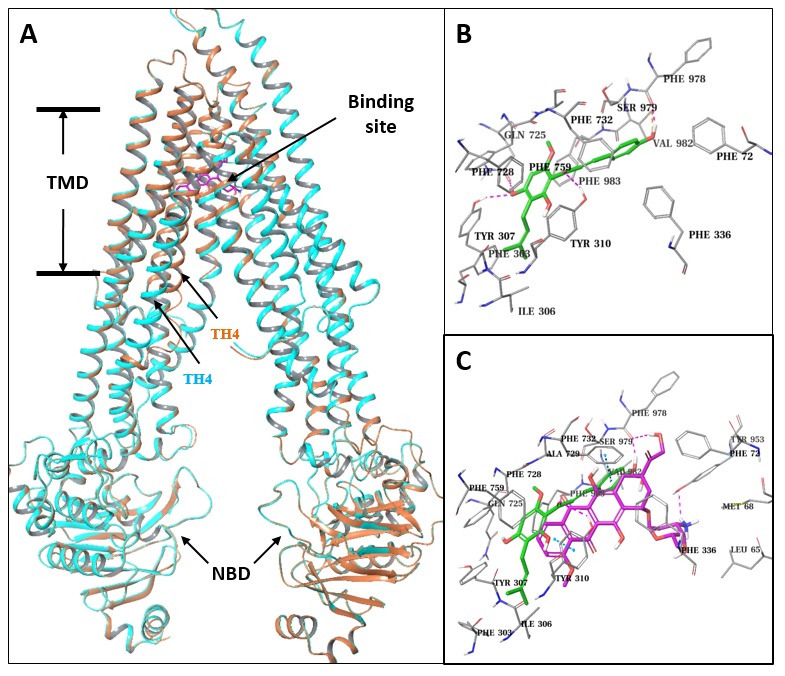
Professor Ming liu, School of Medicine and Pharmacy, Ocean University of China Prenylflavonoid Isoxanthohumol Sensitizes MCF-7/ADR Cells to Doxorubicin Cytotoxicity Isoxanthohumol is a unique prenylflavonoid with the highest content in beer. Isoxanthohumol has multiple bioactivities and has recently received considerable attention inthe scientific community. Nonetheless; its effect on drug resistant cancer cells has rarely been studied. In this paper; we investigated the synergistic effect of isoxanthohumol and doxorubicin on doxorubicin resistant MCF-7/ADR cells. Our results showed that isoxanthohumol sensitizedthe cytotoxic effect of doxorubicin on MCF-7/ADR cells via increased proliferation inhibition and apoptosis stimulation. Molecular mechanism studies further demonstrated that isoxanthohumolinhibited ABCB1-mediated doxorubicin efflux; stimulated the ATPase activity of ABCB1 (ATP-bindingcassette sub-family B member 1); and acted as an ABCB1 substrate. Molecular docking resultssuggested that isoxanthohumol bound to the central transmembrane domain of ABCB1 and itsbinding site overlapped with the doxorubicin binding site. The present studies demonstrated thatisoxanthohumol was a competitive ABCB1 inhibitor which reversed ABCB1-mediated doxorubicinresistance in MCF-7/ADR cells; and therefore could be further developed to help with overcoming ABCB1-mediated drug resistance.
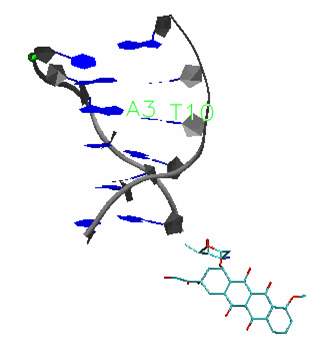
Professor Yujun Zheng, School of physics, Shandong University, Jinan 250100, China Binding of Anticancer Drug Daunomycin to a TGGGGT G-Quadruplex DNA Probed by All-Atom Molecular Dynamics Simulations: Additional Pure Groove Binding Mode and Implications on Designing More Selective G-Quadruplex Ligands
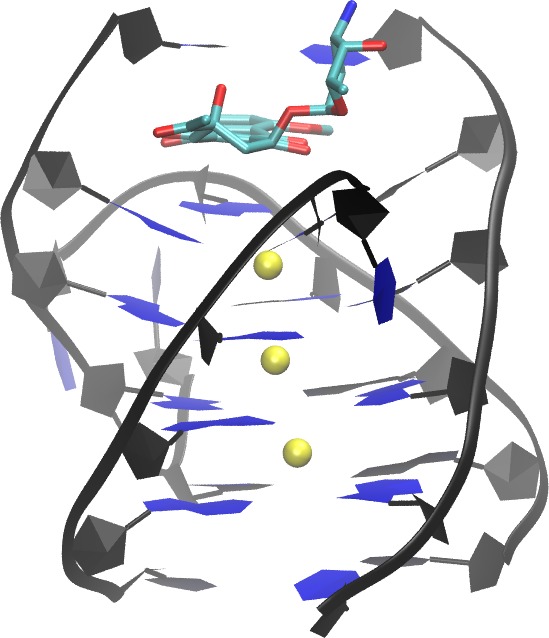
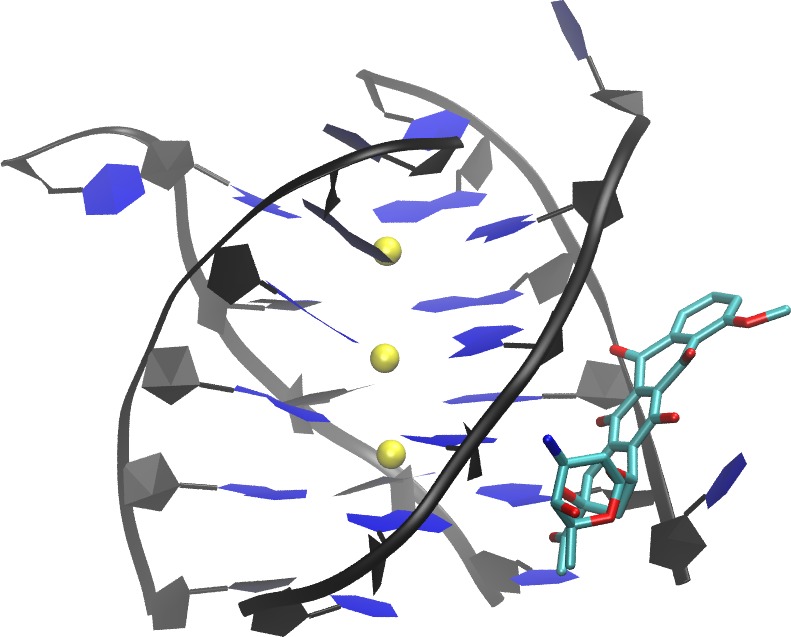
A G-quadruplex structures are emerging cancer-specific targets for chemotherapeutics. Ligands that bind to and stabilize DNA G-quadruplexes have potential to be anti-cancer drugs. Lack of binding selectivity to DNA G-quadruplex over DNA duplex remains a major challenge when attempting to develop G-quadruplex ligands into successful anti-cancer drugs. Thorough understanding of the binding nature of existing non-selective ligands that bind to both DNA quadruplex and DNA duplex will help to address this challenge. Daunomycin and doxorubicin, two commonly used anticancer drugs, are examples of non-selective DNA ligands. In this study, we extended our early all-atom binding simulation studies between doxorubicin and a DNA duplex (d(CGATCG)2) to probe the binding between daunomycin and a parallel DNA quadruplex (d(TGGGGT)4 ) and DNA duplex. In addition to the end stacking mode, which mimics the mode in the crystal structure, a pure groove binding mode was observed in our free binding simulations. The dynamic and energetic properties of these two binding modes are thoroughly examined, and a detailed comparison is made between DNA quadruplex binding modes and DNA duplex binding modes. Implications on the design of more selective DNA quadruplex ligands are also discussed Publication: Shen, Z.; Mulholland, K.; Zheng, Y.; Wu, C.*, Binding of Anticancer Drug Daunomycin to a TGGGGT G-Quadruplex DNA Probed by All-Atom Molecular Dynamics Simulations: Additional Pure Groove Binding Mode and Implications on Designing More Selective G-Quadruplex Ligands. Journal of Molecular Modeling 2017, 23:256
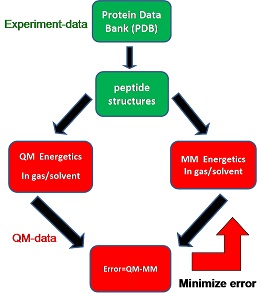
Professor Zhixiang Wang Graduate University of Chinese Academy of Sciences Development of molecular mechanism protein and lipid force field Recently, we have developed a tetrapeptide structure database1 for benchmarking and optimizing protein force fields. The conformations of the tetrapeptides are defined using the most populated structural values from the protein structure database (PDB), which enables us focus on the key conformations and avoid the conformation complexity problem. Next, the high level quantum mechanics (QM) energetics with implicit solvent (MP2/cc-pVTZ//IEFPCM/UAKS2) on the tetrapeptide structures is used as reference data to rank existing force field/ continuum solvent models and a new version implicit protein force field is being developed by minimizing the errors between the molecular mechanics (MM) energetics and the QM energetics using (Fig. 1). Our preliminary results show 20% error reduction of this new protein force field compared with ff03. We will continue our effects in the future.
Publications 1. Jiang JL, Wu YB, Wang ZX*, Wu C* (2010) “Assessing the Performance of Popular Quantum Mechanics and Molecular Mechanics Methods and Revealing the Sequence-Dependent Energetic Features Using 100 Tetrapeptide Models.” J Chem Theory Comput 6: 1199-1209 2. Wen MW, Jiang JL, Wang ZX*, Wu C* (2014) “How accurate are continuum solvation models for amino acid side-chain analogs?” Theor Chem Acc 133:1471
Movie (download and play):
(Phase II drug for Alzheimer's disease) Folding of FSD-1 (a+b) protein
(Predict correct structure from sequence)
Binding of PIB to Ab42 protofibrils
(Predict binding site) Binding of Cancer drug to DNA Quadruplet
(Predicted new binding mode to kill cancer cells)
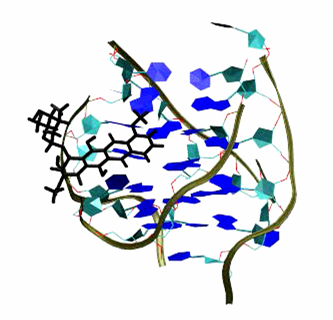
Binding of Cancer drug to DNA Duplet
(Predicted new insertion mode)
(Function bending mode)
Open-close motion of Insulin Degrading Enzyme (IDE)
(Open "mouse" and digest our enemies amylin, Abeta peptides )
Insertion of Adrenaline (Agonist) into beta-Adrenergic receptor (GPCR)
(Signal transduction)
|
|||||||||||
This site was last updated 05/26/25